MACHADO-JOSEPH DISEASE - Treatment in Lisbon
Specialised assessment and follow-up for Machado-Joseph Disease (SCA3) in Lisbon
WHAT IS MACHADO-JOSEPH DISEASE?

The most common spinocerebellar ataxia in the world
Machado-Joseph disease, also known as spinocerebellar ataxia type 3 (SCA3), is an autosomal dominant inherited neurological disorder caused by the expansion of CAG repeats in the gene ATXN3. It is the most prevalent form of genetically determined spinocerebellar ataxia worldwide, with a particular incidence in Portugal - especially in the Azores islands, where its original description gave the disease its name.
Most frequent clinical manifestations
- Progressive cerebellar ataxiaGait instability, limb dysmetria (differences in length) and dysarthria (difficulty articulating speech), which evolve slowly and are not always obvious.
- Ophthalmoplegia and nystagmusChanges in eye movements, double vision (diplopia) and difficulty fixing moving objects.
- Spasticity and pyramidal signsMuscle stiffness, involuntary movements and frequent painful spasms in the lower limbs.
- Peripheral neuropathy and sleep disordersDecreased sensitivity, discomfort in the legs (especially at night) and sleep disturbances impair rest and quality of life.
CLINICAL FORMS AND CHARACTERISTICS OF DMJ
Type I - Pyramidal and Extrapyramidal Form
It starts earlier (before the age of 30) and causes muscle stiffness, spasticity (tense muscles) and difficulty in movement. It usually progresses more quickly.
Early onsetType II - Classic Cerebellar Form
This is the most common form. It starts between the ages of 20 and 50 and causes lack of coordination, difficulty speaking and changes in eye movements, with a progressive worsening of balance when walking.
Most frequent formType III - Form with Peripheral Neuropathy
It starts later in life (after the age of 40) and causes lack of coordination, weakness and loss of muscle mass, as well as small involuntary muscle movements.
Late onsetGenetic basis - CAG expansion in ATXN3
The disease is caused by a genetic alteration in the gene ATXN3. The greater this alteration, the earlier the symptoms appear and the greater the severity of the disease tends to be.
Autosomal dominant
Portugal - particularly the Azores - has one of the highest prevalences of JDM in the world, making early diagnosis a national clinical priority.
MONITORING AND THERAPEUTIC IMPACT
IN MACHADO-JOSEPH DISEASE
Early multidisciplinary intervention improves quality of life and delays functional progression
* Data based on published clinical studies and patient records. Individual results may vary.
Sources: clinical data, NIH/PubMed - Sequeiros & Coutinho, global prevalence SCA3/DMJ (25-50% of SCAs); NIH/PubMed - meta-analysis of physiotherapy in degenerative cerebellar ataxias (SARA MD = -1.41); Azevedo CJ et al. - Prevalence of MJD in the Azores, 39/100,000 (JSciMedCentral); NIH/PubMed - Riess O. et al., SCA3 represents 58% of the SCAs in Portugal.




IMPORTANCE OF SPECIALISED CONSULTATION
Diagnosing Machado-Joseph Disease requires clinical experience in recognising its forms of presentation, as well as access to genetic confirmation and structured neurological assessment protocols. Many patients go through years of research without a definitive diagnosis - especially when there is no known family history or when the initial symptoms are subtle. Ideally, if there is suspicion, you should seek out a specialist in this condition specifically.
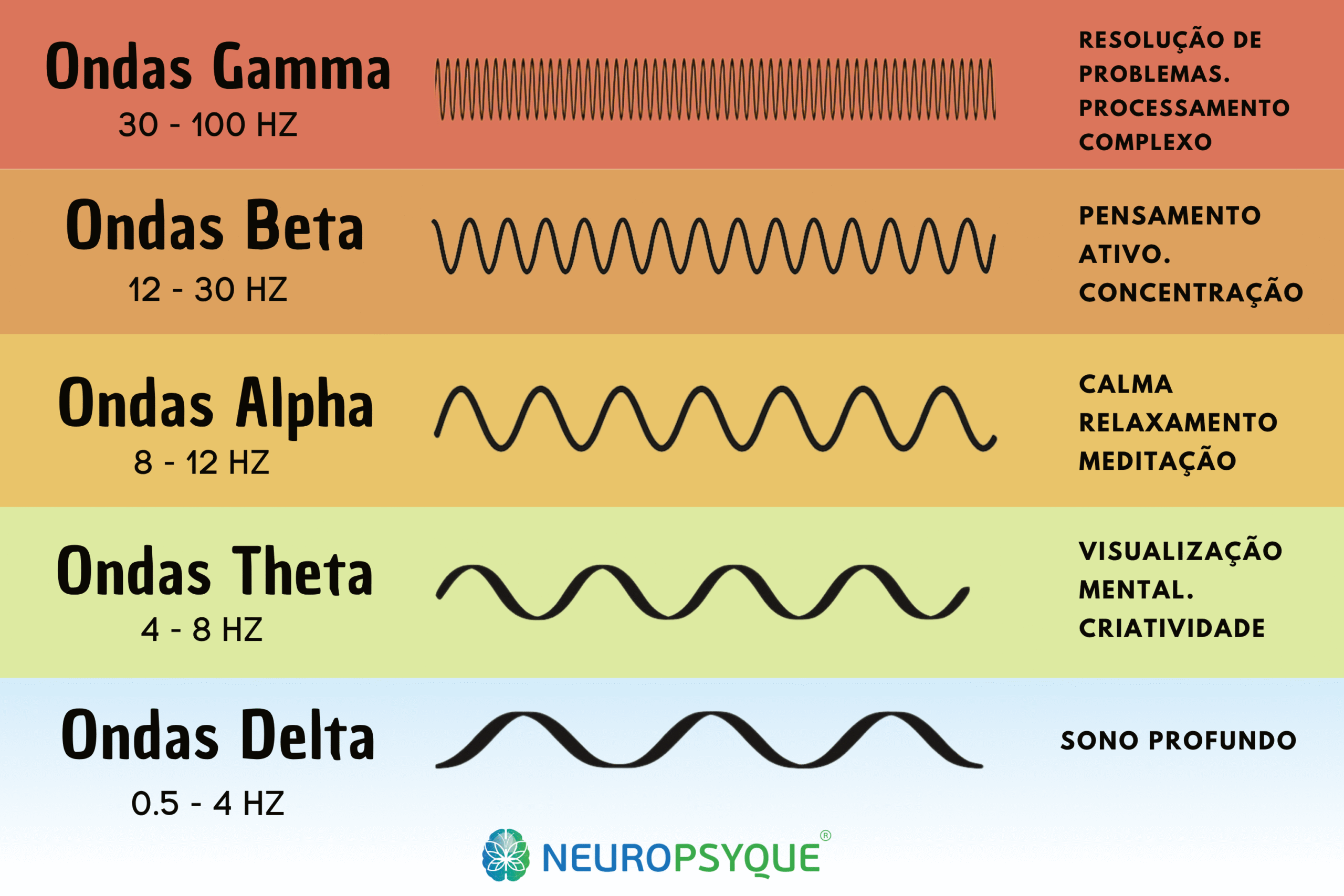
At Clínica NeuroPsyque we monitor different types of ataxia, but you should look for parallel monitoring specialised in the condition. The follow-up of JDM is multidisciplinary and longitudinal: periodic neurological assessment, non-invasive neuromodulation to control spasticity and associated symptoms, support in cognitive and motor rehabilitation, and liaison with the family doctor and other specialists. The aim is to preserve functional autonomy for as long as possible - with scientific rigour and close human support.